




Unlocking Precision Medicine in Multiple Myeloma: Venetoclax and Novel Targeted Therapies

Abstract
Precision medicine, as defined by individualized therapeutic targeting of oncogenic mutations, has had limited success in treating Multiple Myeloma (MM). FISH cytogenetics and next generation sequencing (NGS) have led to attempts of subtype classification, however this minimally affects treatment decisions to date. Additionally, identifying accurate and clinically available biomarkers to signal drug sensitivities has proven difficult for MM. This review discusses the previous attempts at precision medicine in MM with an in-depth focus on BCL2 inhibition. Next, select ongoing studies are reviewed including discussing relevant biomarkers being utilized in current clinical trials. Finally, we discuss potential future directions for a promising treatment approach.
Key Words: Multiple Myeloma, Precision Medicine, Personalized Medicine, Targeted Therapy, Plasma Cell Malignancies, Venetoclax
Introduction
Precision medicine harnesses a patient’s genomic data to identify and attack tumor-specific vulnerabilities with personalized therapies. This strategy requires the right treatment and a reliable biomarker to identify cases that will respond. There is no better example of the success of precision medicine than in hematologic malignancies, e.g., IDH1/2 and FLT3 inhibitors in acute myeloid leukemia (AML), BCR-ABL1 inhibitors for chronic myelogenous leukemia (CML), and BTK inhibitors in non-Hodgkin lymphoma (NHL), to name a few. Multiple myeloma (MM), however, has not shared in such success. To date, effective therapies in MM have targeted vulnerabilities in plasma cell biology, such as high protein turnover or overexpression of specific cell markers. Less success has been had in targeting genetic subtypes of MM.
MM has historically been regarded as a single disease despite a variety of clinical presentations and treatment outcomes. Although many patients with standard-risk myeloma achieve nearly 10 years of event-free survival after frontline therapy, ultra-high-risk patients may relapse within 12 months1. This variation in clinical response suggests that there are subgroups of myeloma that may warrant individualized treatment. The difficulty in developing precision medicine strategies for MM may be due to the extreme heterogeneity between MM cases2,3,4. Even within a single case of MM, molecular events occur in a branching pattern resulting in intra-clonal heterogeneity leading to subclones with novel mutations that may influence drug sensitivity5,6. In the face of such genetic complexity, little progress has been made in the therapeutic targeting of oncogenic mutations in MM. Instead, MM is currently treated like one disease, albeit with some variation due to clinical variables like age and fitness. FISH cytogenetics and next-generation sequencing (NGS) have led to attempts of subtype classification, however this minimally affects treatment decisions to date. Additionally, utilizing biomarkers to signal potential drug sensitivities has proved difficult7. This narrative review describes the previous attempts, ongoing studies, and future directions of precision medicine with targeted therapies in MM. A literature search was conducted in PubMed and Web of Science for articles using the search terms “precision medicine” “multiple myeloma” “targeted therapy” BCL2” “NGS” and related keywords were used. Findings were synthesized thematically to highlight targeted therapy trends in multiple myeloma drug development.
BCL2 and t(11;14)
The t(11;14)(q13;q32) is the most common chromosomal translocation in MM occurring in 15-20% of cases8. The translocation juxtaposes the cyclin D1 (CCND1) gene with immunoglobulin heavy chain enhancer elements leading to cyclin D1 overexpression promoting cell cycle progression9.However, an understanding of the full oncogenic functions is incomplete. MM cells with t(11;14) can have unique clinical features such as lymphoplasmacytoid morphology, CD20 expression, and oligosecretory or free light chain only monoclonal protein production10,11,12. MM cells with t(11;14) have also been found to have a unique balance of BCL2 family of proteins13.
The B-cell lymphoma-2 (BCL2) gene was so named from its original identification as the portion of chromosome 18 involved in the t(14;18)(q32;q21) occurring in over 80% of follicular lymphomas14. Subsequently multiple proteins were identified in the BCL2 protein family to include both anti-apoptotic and pro-apoptotic proteins. In many hematologic neoplasms, the overexpression of anti-apoptotic BCL2 proteins is an important factor in survival and drug resistance15. The malignant plasma cells of MM express at least 3 anti-apoptotic BCL2 family members: BCL2, B-cell lymphoma extra-large (Bcl-xL), and myeloid cell leukemia-1 (MCL1)16. Most normal plasma cells and most MM cells carry out anti-apoptotic signaling primarily through MCL1 and are occasionally codependent on Bcl-xL or BCL217. However, t(11;14) MM cells tend to have a predominant dependence on BCL2 and a lower MCL1 expression resulting in a high BCL2 to MCL1 ratio, albeit BCL2 dependence may exist in other myeloma subgroups as well Figure 1 16. In preliminary studies of novel BCL2 inhibitors, cell death was demonstrated in the sub-group of myeloma with a high BCL2/MCL1 ratio18.
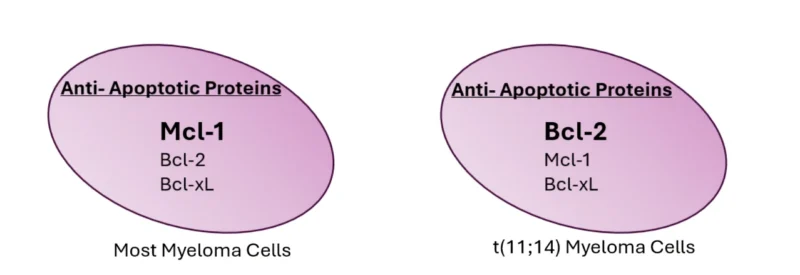
Figure 1: Most myeloma cells rely on MCL1 for anti-apoptotic signaling. Myeloma cells harboring a t(11;14) tend to have a predominant dependence on BCL2 and lower MCL1 expression.
Venetoclax was the first selective BCL2 inhibitor utilized in clinical practice. A phase 1 study of venetoclax in relapsed or refractory MM (RRMM) showed that high expression of BCL2 relative to MCL1 was associated with improved response to venetoclax monotherapy19. However, to overcome MCL1 activity, preclinical studies suggested that dexamethasone and proteasome inhibitors could shift the reliance on MCL1 to BCL2 in MM cells without a t(11;14)20,21. The synergy seen with the combination of venetoclax and bortezomib preclinically provided a rationale to evaluate the combination of venetoclax, bortezomib, and dexamethasone in all MM patients, not just those with high BCL2 expression or t(11;14). The initial phase 1 study evaluated the combination of venetoclax with bortezomib and dexamethasone in 66 RRMM patients. The study showed acceptable safety with an overall response rate (ORR) of 67% with only 14% of patients having a known t(11;14)22. These encouraging results led to the phase 3 BELLINI trial which randomized RRMM patients in a 2:1 fashion to venetoclax, bortezomib, dexamethasone compared to bortezomib dexamethasone. Patients had 1 to 3 prior lines of therapy and were still sensitive to proteasome inhibitors23. The primary endpoint was progression-free survival (PFS). Median PFS was significantly longer in the venetoclax group (22.4 months 95% CI 15.3 – NE) than in the placebo group (11.5 months 95% CI 9.6 – 15). However, the study was halted early due to increased deaths in the venetoclax arm and a decreased overall survival trend. Such a detriment was not seen in the subgroup of patients harboring a t(11;14) therefore further evaluation of venetoclax was focused on the subgroup of patients with t(11;14)24.
A phase 1/2 study treated only patients with a t(11;14) RRMM with venetoclax and dexamethasone. In the phase 2 cohort, patients had a median of 5 prior lines of therapy and achieved an ORR of 48%25. A phase 3 study was then developed, the CANOVA trial, which enrolled only RRMM patients with a t(11;14) and > 2 prior lines of therapy randomizing patients to venetoclax and dexamethasone or pomalidomide and dexamethasone. Despite selecting only patients with t(11;14), the end results did not demonstrate significant improvement in the venetoclax dexamethasone arm. The prolonged median PFS of 9.9 months in the venetoclax arm was numerically longer than the 5.8 months in the pomalidomide arm but the results did not meet statistical significance (HR 0.823, 95% CI: 0.596 – 1.136, p: 0.237). The ORR was higher in the venetoclax arm (62% vs. 35%) and the median OS was longer in the venetoclax arm (32.4 months vs. 24.5 months)(HR 0.697, 95% CI: 0.472 – 1.029, nominal p 0.067). Cross-over events were not reported. The median time to next treatment was longer in the venetoclax arm (21.2 months vs. 8.3 months)(HR of 0.546 (95% CI: 0.385- 0.776); nominal p-value of 0.001)26. A post-hoc analysis evaluating the responses in patients with BCL2 high gene expression by RNAseq was conducted. The analysis suggested that the ORR was higher in patients with high BCL2 expression (3.7 log2 FPKM cutoff), however the median PFS and median OS did not differ between groups27.
The story of venetoclax in MM identifies a common challenge in precision medicine of requiring an accurate biomarker to predict response. While both t(11;14) and BCL2 expression may be a reasonable avenue to identify patients that will respond to BCL2 inhibition, clinical experience has proven to provide inconsistent results. In the case of BCL2 expression, the co-expression of other anti-apoptotic proteins such as MCL1 may limit the utility of BCL2 IHC expression alone28.
However, the attempt at utilizing t(11;14) and/or BCL2 as an avenue for precision medicine in MM is far from over. An ongoing Phase 2 study is investigating the addition of venetoclax to carfilzomib and dex (Kd) in t(11;14) positive patients. An abstract in 2023 reported a 92% ORR in the venetoclax Kd arm compared to 63% in the Kd arm29. Additional selective BCL2 inhibitors are being investigated as well, such as sonrotoclax and lisaftoclax. Sonrotoclax inhibits BCL2 at lower concentrations compared to venetoclax in both biochemical and cellular assays. BGB-11417-105 is an ongoing open-label phase 1b/2 dose escalation study of sonrotoclax in patients with a t(11;14) RRMM. As of March 25th, 2024, a total of 32 patients have been treated at the recommended dose for expansion (RDFE) of 640 mg in combination with dexamethasone. Among 24 evaluable patients, the ORR was 75% (95% CI, 53-90%)30. Lisaftoclax (APG-2575) is another selective BCL2 being evaluated with other treatment combinations in RRMM, not restricted to t(11;14)31. Results from these studies are eagerly awaited.
MCL1 and 1q+
Other studies are attempting to circumvent the intrinsic venetoclax resistance by adding additional inhibitors. As discussed above, most myeloma cells rely on MCL1 for anti-apoptotic properties thus preventing the reliance on BCL2. Additionally, MCL1 dependency increases from diagnosis to relapse as overexpression of MCL1 is detected in 52% of patients at diagnosis and 81% at relapse32. MCL1 is a larger protein than BCL2 (40 kDa vs. 26 kDa) and has been indicated in myeloma development32,33 and venetoclax resistance34. MCL1 resides on chromosome 1q2133,35 which is a common amplification or gain found in MM patients associated with reduced survival36. Therefore, MCL1 has been an interesting potential target for MM. Early studies are starting to look into MCL1 inhibitors such as tapotoclax (AMG176), AZD599137, KS1838 and S63845 however there has been limited clinical success in other hematologic malignancies to date39. A particular challenge has been cardiotoxicity, however MCL1 inhibitors with a reduced half-life are trying to circumvent this issue40,41. Attempts at evading venetoclax resistance have not stopped at the BCL2 family of proteins either. A recent preclinical study by Okamoto et al. showed that an RSK/AKT/S6K inhibitor could induce synergistic apoptosis with venetoclax42. Such novel therapeutic avenues have not made it into clinical studies to date.
NRAS/KRAS Mutations
Oncogenic RAS signaling is among the most common oncogenic drivers in human cancer and can be found in 40-60% of MM tumors2,3,43. The RAS family, comprised of KRAS, NRAS, HRAS are small GTPases that act as molecular switches regulating downstream signaling such as the mitogen-activated protein kinase (MAPK) pathway, the phosphoinositide 3-kinase (PI3-K) or mTORC1 pathways. Within the MAPK pathway, RAS mutations lead to downstream MEK activation which enhances survival, proliferation, and migration of MM cells. RAS itself has been historically difficult to target, so focus has been made on the downstream effectors. When constitutively active, both pathways may lead to uncontrolled cell growth. The incidence of RAS mutations goes up with more advanced/relapsed disease suggesting a role in clonal evolution and treatment resistance3. Preclinical evidence suggests that MAPK activation plays a role in IMiD resistance and MEK inhibition may reinstate IMiD sensitivity44.
Single-agent MEK inhibition has been explored in MM in two small trials. One trial evaluated selumetinib in MM patients unselected for RAS mutations and resulted in a 5.6% overall response rate (NCT01085214)45. Another study treated KRAS, NRAS, or BRAF mutant MM patients with trametinib and, despite selecting for RAS mutant disease, only 1 of 12 patients achieved a response (NCT01907815)46. Therefore, even in patients with a known RAS mutation, single-agent activity is minimal at best.
Combination therapy involving MEK inhibitors has also been explored in MM. One trial has evaluated MEK inhibition cobimetinib +/- venetoclax +/- atezolizumab (NCT03312530)47. There were no responses seen in the cobimetinib single agent arm despite having about 50% of the patients with RAS mutations. There were some responses in the cobimetinib + venetoclax arms but in patients with t(11;14) so the activity could have been primarily due to venetoclax. Trametinib is also being explored in combination with mezigdomide in the ongoing Phase 1/2 CA057-003 Trial (cohort C). Preliminary results from the efficacy-evaluable population showed a 75% ORR48.
Recent preclinical data suggests that the inhibition of both MEK and the mammalian Target of Rapamycin (mTORC) pathways is needed to impede RAS-dependent pathogenic signaling in MM cells49. In KRAS or NRAS dependent cell lines, mTORC1 was found to be active downstream of RAS. However, inhibition of mTORC1 alone enhanced reliance on MEK signaling. Therefore, MEK and mTOR inhibition were required to halt oncogenic signaling. A new study will be exploring the efficacy and safety of combining a MEK inhibitor with an mTOR inhibitor (NCT06876142).
In parallel with pathway-targeted approaches, immunotherapeutic strategies have also been explored to directly target RAS-mutated myeloma. TG01, a peptide-based vaccine composed of mutant RAS epitopes, has been evaluated in a phase I/II study in patients with relapsed/refractory MM or high-risk smoldering MM harboring KRAS or NRAS mutations. Preliminary results reported that the vaccination led to increased TG01-specific T-cell responses in approximately one-third of evaluable patients, although no objective clinical responses have yet been observed50.
BRAF V600E Mutation
BRAF mutations are another targetable mutation found in myeloma. While less frequent than RAS, BRAF V600E mutations can be found in 4 to 8% of RRMM patients51. In melanoma and hairy cell leukemia targeting BRAF V600E with a combination of a MEK inhibitor and BRAF inhibitor is more effective than using a BRAF inhibitor alone. Stemming from that principal, a combination approach with a MEK inhibitor (binimetinib) and a BRAF inhibitor (encorafenib) was evaluated in a Phase 2 study for patients with RRMM and BRAF V600E mutations (NCT02834364)52. Out of 12 patients treated, the ORR was 83.3%, suggesting merit in a targeted precision medicine approach in MM. Interestingly in this study, the authors note that despite the high ORR, patients relapsed quickly, and emerging resistance to therapy was driven by RAS mutations.
t(4;14): MMSET and FGFR3
The t(4;14) is the second most common translocation found in 10-15% of patients with MM53,54. This translocation can result in the simultaneous stimulation of two oncogenes: the fibroblast growth factor receptor 3 (FGFR3) gene and the multiple myeloma SET domain containing protein (MMSET) (also known as the nuclear receptor SET domain protein 2 – NSD2) gene. This is a unique example of an IgH translocation stimulating two oncogenic genes on reciprocal translocations55. On the derivative chromosome 4 [der(4)], the transcription of the MMSET domain comes under the control of the IgH super enhancer. On the derivative chromosome 14 [der(14)], an IgH enhancer stimulates the transcription of the FGFR3 locus (Figure 2).
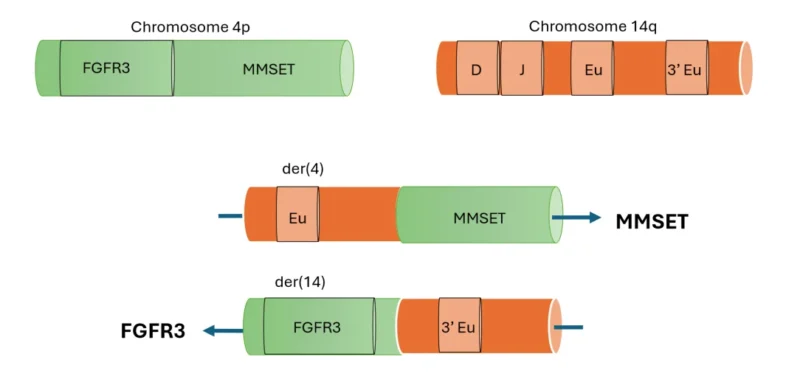
Figure 2: The t(4;14) translocation results in stimulating two oncogenic genes on reciprocal translocations. On the derivative chromosome 4 {der(4)], the transcription of the MMSET domain becomes under control of the IgH super enhancer. On the derivative chromosome 14 [der(14)], an IgH enhancer stimulates the transcription of the FGFR3 locus.
FGFR3 (fibroblast growth factor receptor 3), MMSET (multiple myeloma SET domain containing protein) Eu (IgH enhancer)
MMSET is a histone 3 lysine 36 methyltransferase which becomes overexpressed and leads to the transcription of pro-cancer genes. KTX1001 is an oral inhibitor of MMSET (multiple myeloma SET domain containing protein) that is under investigation for patients with t(4;14) (NCT05651932).
FGFR-3 is a growth factor receptor that can activate RAS signaling and is frequently overexpressed in MM due to t(4;14) and rarely gene amplifications54,56. Erdafitinib is a small molecule inhibitor of the fibroblast growth factor receptor (FGFR) which is a tyrosine kinase. Erdafitinib is currently FDA approved for bladder cancer with an FGFR3 or FGFR2 alteration. A trial was initiated evaluating erdafitinib with dexamethasone in RRMM but was terminated due to low patient accrual (NCT02952573). The study investigators did report on one patient with IgA lambda t(4:14) positive RRMM who, despite clinically progressing, was found to have elimination of the FGFR3-mutant subclone after treatment. The patient’s clinical progression was due to the expansion of a preexisting clone with loss of chromosome 17p57. A story that once again emphasizes the challenging heterogenous sub-clonal nature of MM.
Utilizing an Umbrella Trial for Genomically Guided Therapies
The Myeloma Developing Regimens Using Genomics (MyDRUG) study is a multicenter, umbrella-style precision medicine trial designed to evaluate genomically guided therapies in patients with functionally high-risk multiple myeloma (MM) (NCT03732703). Functionally high-risk disease is defined by early relapse following frontline therapy, occurring within three years after autologous stem cell transplantation or within eighteen months in patients who did not undergo transplantation. The study aims to determine whether targeting dominant genomic alterations earlier in the disease course can improve outcomes in this high-risk population.
MyDRUG assigns patients to molecularly defined treatment arms based on the presence of a ≥30% mutational burden in predefined genomic alterations. Each arm combines a targeted agent with a common myeloma backbone of ixazomib, pomalidomide, and dexamethasone. The six investigational cohorts include: CDKN2C deletion treated with abemaciclib; FGFR3 alterations treated with erdafitinib; KRAS or NRAS mutations treated with cobimetinib; BRAF V600E mutations treated with cobimetinib; IDH2 mutations treated with enasidenib; and t(11;14) translocation treated with venetoclax (Table 1).
Table 1: MyDRUG Trial Arms
| Genomic Alteration (≥30% Mutational Burden) | Targeted Agent | Mechanism of Action | Combination Backbone | Reported Clinical Activity |
|---|---|---|---|---|
| CDKN2C deletion | Abemaciclib | CDK4/6 inhibitor | Ixazomib, pomalidomide, dexamethasone | Not yet reported |
| FGFR3 alteration | Erdafitinib | FGFR tyrosine kinase inhibitor | Ixazomib, pomalidomide, dexamethasone | Not yet reported |
| KRAS / NRAS mutation | Cobimetinib | MEK inhibitor | Ixazomib, pomalidomide, dexamethasone | ORR 86% (6/7 patients); 4 PR, 2 VGPR (ASCO 2022) |
| BRAF V600E mutation | Cobimetinib | MEK inhibitor | Ixazomib, pomalidomide, dexamethasone | Not yet reported |
| IDH2 mutation | Enasidenib | IDH2 inhibitor | Ixazomib, pomalidomide, dexamethasone | Not yet reported |
| t(11;14) translocation | Venetoclax | BCL-2 inhibitor | Ixazomib, pomalidomide, dexamethasone | Not yet reported (activity extrapolated from venetoclax studies) |
Preliminary clinical activity has been reported from the RAS-mutated cohort. An abstract presented at the 2022 ASCO Annual Meeting described outcomes in seven patients with a median of one prior line of therapy58. Six patients achieved a response, including four partial responses and two very good partial responses. While these findings suggest promising activity, interpretation is limited by the small sample size and single-arm design, making it difficult to isolate the contribution of the targeted agent beyond the active backbone regimen. Nonetheless, these early efficacy and safety signals support continued investigation and provide a rationale for future randomized studies.
Conclusions
Despite many valiant efforts, currently there are no FDA approved therapies for molecularly defined subsets of multiple myeloma. Numerous precision medicine strategies have been evaluated in the relapsed and refractory setting as discussed above, and their limited success underscores the biological complexity and clonal heterogeneity of advanced disease. Precision medicine strategies may ultimately prove more effective when deployed earlier in the disease course, before extensive clonal evolution and therapeutic resistance have occurred. This review focused on targeted therapies as precision medicine, however other strategies, such as tailoring treatment to disease response or minimal residual disease, is another such example of precision medicine in MM59. Nevertheless, encouraging signals, such as the improved PFS with venetoclax in t(11;14) patients or the high ORR with encorafenib and binimetinib in BRAF V600E mutated myeloma provide a glimmer of hope that molecularly targeted approaches may yet play a role in selected patients. Potentially the correct biomarker is not genetically based but found via gene expression or proteomic profiling. Evaluating protein expression and signaling states can distinguish MM subtypes that look similar genomically but behave very differently clinically. Tools like phosphoproteomics can identify active kinases even when no obvious mutation is present60. This may help explain why patients with the same cytogenetic risk can respond very differently to the same therapy. Regardless of the biomarker discovered, we need to find ways to overcome the resistance that can quickly occur to targeted therapy. MM remains incurable and there remains a need for novel treatment approaches. Precision medicine may hold the key to cure, but success will likely require the correct combination of multiple targeted agents and ability to select the right patient at the right time. Undoubtedly, for targeted therapies to work in relapsed refractory MM, a multiagent personalized approach will be needed.
Conflict of Interest: The authors declare that there is no conflict of interest.
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
License
© The Author(s) 2026.
This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, and unrestricted adaptation and reuse, including for commercial purposes, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. To view a copy of this license, visit https://creativecommons.org/licenses/by/4.0/.
References
-
Martino EA, Mele G, Vigna E, Morabito F, Gentile M. Refining High-Risk Multiple Myeloma: Advancements in Genomic, Clinical, and Prognostic Criteria. Mediterr J Hematol Infect Dis. 2025 Jan 1;17(1):e2025006.
-
Kortüm KM, Mai EK, Hanafiah NH, et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood. 2016 Sep 1;128(9):1226-33.
-
Walker BA, Mavrommatis K, Wardell CP, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood. 2018 Aug 9;132(6):587-597.
-
Brioli A, Melchor L, Cavo M, Morgan GJ. The impact of intra-clonal heterogeneity on the treatment of multiple myeloma. Br J Haematol. 2014 May;165(4):441-54.
-
Lohr JG, Stojanov P, Carter SL, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014 Jan 13;25(1):91-101.
-
Tirier SM, Mallm JP, Steiger S, et al. Subclone-specific microenvironmental impact and drug response in refractory multiple myeloma revealed by single-cell transcriptomics. Nat Commun. 2021 Nov 29;12(1):6960.
-
Cani L, Gupta VA, Kaufman JL. BCL2 inhibition for multiple myeloma and AL amyloidosis. Br J Haematol. 2025 May;206(5):1285-1296.
-
Avet-Loiseau H, Attal M, Moreau P, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myélome. Blood. 2007 Apr 15;109(8):3489-95
-
Chesi M, Bergsagel PL, Brents LA, Smith CM, Gerhard DS, Kuehl WM. Dysregulation of cyclin D1 by translocation into an IgH gamma switch region in two multiple myeloma cell lines. Blood. 1996 Jul 15;88(2):674-81.
-
Robillard N, Avet-Loiseau H, Garand R, et al. CD20 is associated with a small mature plasma cell morphology and t(11;14) in multiple myeloma. Blood. 2003 Aug 1;102(3):1070-1.
-
El Hussein S, Medeiros LJ, Hu S, Lin P, Wang W. The many faces of plasma cell neoplasms: morphological and immunophenotypical variants of the great imitator. Pathology. 2022 Feb;54(1):32-42.
-
Amy Heerema-McKenney, James Waldron, Steven Hughes, et al. Shaughnessy, Clinical, Immunophenotypic, and Genetic Characterization of Small Lymphocyte–Like Plasma Cell Myeloma: A Potential Mimic of Mature B-Cell Lymphoma. American Journal of Clinical Pathology. Volume 133, Issue 2, February 2010, Pages 265–27.
-
Paner A, Patel P, Dhakal B. The evolving role of translocation t(11;14) in the biology, prognosis, and management of multiple myeloma. Blood Rev. 2020 May;41:100643.
-
Cleary ML, Sklar J. Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proc Natl Acad Sci U S A. 1985 Nov;82(21):7439-43.
-
Pettersson M, Jernberg-Wiklund H, Larsson LG, Sundström C, Givol I, Tsujimoto Y, Nilsson K. Expression of the bcl-2 gene in human multiple myeloma cell lines and normal plasma cells. Blood. 1992 Jan 15;79(2):495-502.
-
Touzeau C, Ryan J, Guerriero J, et al. BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia. 2016 Mar;30(3):761-4.
-
Derenne S, Monia B, Dean NM. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood. 2002 Jul 1;100(1):194-9.
-
Touzeau, C., Dousset, C., Le Gouill, S. et al. The Bcl-2 specific BH3 mimetic ABT-199: a promising targeted therapy for t(11;14) multiple myeloma. Leukemia. 2014. 28(1): p. 210-212.
-
Kumar S, Kaufman JL, Gasparetto C, et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood. 2017 Nov 30;130(22):2401-2409.
-
Matulis SM, Gupta VA, Nooka AK, et al.Dexamethasone treatment promotes Bcl-2 dependence in multiple myeloma resulting in sensitivity to venetoclax. Leukemia. 2016 May;30(5):1086-93.
-
Punnoose EA, Leverson JD, Peale F, et al. Expression Profile of BCL-2, BCL-XL, and MCL-1 Predicts Pharmacological Response to the BCL-2 Selective Antagonist Venetoclax in Multiple Myeloma Models. Mol Cancer Ther. 2016 May;15(5):1132-44.
-
Moreau P, Chanan-Khan A, Roberts AW, et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood. 2017 Nov 30;130(22):2392-2400.
-
Kumar SK, Harrison SJ, Cavo M, et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2020 Dec;21(12):1630-1642.
-
Kumar SK, Harrison SJ, Cavo M, et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in relapsed or refractory multiple myeloma (BELLINI): final overall survival results from a randomised, phase 3 study. Lancet Haematol. 2025 Aug;12(8):e574-e587.
-
Kaufman JL, Gasparetto C, Schjesvold FH, et al. Targeting BCL-2 with venetoclax and dexamethasone in patients with relapsed/refractory t(11;14) multiple myeloma. Am J Hematol. 2021 Apr 1;96(4):418-427.
-
Mateos MV, et al. Results from the randomized, open-label phase 3 CANOVA study of venetoclax–dexamethasone versus pomalidomide–dexamethasone in patients with t(11;14)-positive relapsed/refractory multiple myeloma. Presented at: International Myeloma Society Annual Meeting; 2023.
-
Nizar J. Bahlis, Rakesh Popat, Meral Beksac et al. Efficacy of venetoclax-dexamethasone (VenDex) v pomalidomide-dexamethasone (PomDex) in patients (Pts) with t(11;14)-positive relapsed/refractory multiple myeloma [t(11;14)+ RRMM]: Phase 3 CANOVA study biomarker subgroup analysis.. J Clin Oncol. 42, 7510-7510(2024).
-
Bolarinwa A, Nagaraj M, Zanwar S, et al. Venetoclax-based treatment combinations in relapsed/refractory multiple myeloma: practice patterns and impact of secondary cytogenetic abnormalities on outcomes. Blood Cancer J. 2025 Apr 4;15(1):57.
-
Kaufman JL, Gasparetto C, Kovacsovics TJ, et al. First results from the randomized portion of a phase 2 study of venetoclax plus carfilzomib–dexamethasone vs carfilzomib–dexamethasone in patients with t(11;14) relapsed/refractory multiple myeloma. Presented at: International Myeloma Society Annual Meeting; 2023. Abstract OA-29.
-
Abel Costa L, et al. Sonrotoclax plus dexamethasone was tolerable and demonstrated antimyeloma activity in patients with relapsed/refractory multiple myeloma harboring t(11;14). Presented at: International Myeloma Society Annual Meeting; 2024
-
Sikander Ailawadhi, Asher A. Chanan-Khan, Jack Khouri, et al. Lisaftoclax (APG-2575) Combined with Novel Therapeutic Regimens in Patients (pts) with Relapsed or Refractory Multiple Myeloma (R/R MM) or Immunoglobulin Light-Chain (AL) Amyloidosis. Blood 2024; 144 (Supplement 1): 1022.
-
Wuillème-Toumi S, Robillard N, Gomez P, et al. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia. 2005 Jul;19(7):1248-52.
-
Zhang B, Gojo I, Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood. 2002 Mar 15;99(6):1885-93.
-
Bolomsky A, Miettinen JJ, Malyutina A, et al. Heterogeneous modulation of Bcl-2 family members and drug efflux mediate MCL-1 inhibitor resistance in multiple myeloma. Blood Adv. 2021 Oct 26;5(20):4125-4139.
-
Craig RW, Jabs EW, Zhou P, et al. Human and mouse chromosomal mapping of the myeloid cell leukemia-1 gene: MCL1 maps to human chromosome 1q21, a region that is frequently altered in preneoplastic and neoplastic disease. Genomics. 1994 Sep 15;23(2):457-63.
-
Schmidt TM, Fonseca R, Usmani SZ. Chromosome 1q21 abnormalities in multiple myeloma. Blood Cancer J. 2021 Apr 29;11(4):83.
-
Tron AE, Belmonte MA, Adam A, et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun. 2018 Dec 17;9(1):5341.
-
Al-Odat OS, Elbezanti WO, Gowda K, et al. KS18, a Mcl-1 inhibitor, improves the effectiveness of bortezomib and overcomes resistance in refractory multiple myeloma by triggering intrinsic apoptosis. Front Pharmacol. 2024 Oct 1;15:1436786.
-
Desai P, Lonial S, Cashen A, et al. A Phase 1 First-in-Human Study of the MCL-1 Inhibitor AZD5991 in Patients with Relapsed/Refractory Hematologic Malignancies. Clin Cancer Res. 2024 Nov 1;30(21):4844-4855.
-
Rauh U, Wei G, Serrano-Wu M, et al. BRD-810 is a highly selective MCL1 inhibitor with optimized in vivo clearance and robust efficacy in solid and hematological tumor models. Nat Cancer. 2024 Oct;5(10):1479-1493.
-
Bolomsky A, Vogler M, Köse MC, Heckman CA, Ehx G, Ludwig H, Caers J. MCL-1 inhibitors, fast-lane development of a new class of anti-cancer agents. J Hematol Oncol. 2020 Dec 11;13(1):173.
-
Okamoto H, Mizutani S, Tsukamoto T, et al. Robust anti-myeloma effect of TAS0612, an RSK/AKT/S6K inhibitor, with venetoclax regardless of cytogenetic abnormalities. Leukemia. 2025 Jan;39(1):211-221.
-
Bolomsky A, Young RM. Pathogenic signaling in multiple myeloma. Semin Oncol. 2022 Feb;49(1):27-40.
-
Ocio EM, Fernández-Lázaro D, San-Segundo L, et al. In vivo murine model of acquired resistance in myeloma reveals differential mechanisms for lenalidomide and pomalidomide in combination with dexamethasone. Leukemia. 2015 Mar;29(3):705-14.
-
Holkova B, Zingone A, Kmieciak M, et al. A Phase II Trial of AZD6244 (Selumetinib, ARRY-142886), an Oral MEK1/2 Inhibitor, in Relapsed/Refractory Multiple Myeloma. Clin Cancer Res. 2016 Mar 1;22(5):1067-75.
-
Suzanne Trudel, Nizar J. Bahlis, Christopher P. Venner, et al. Biomarker Driven Phase II Clinical Trial of Trametinib in Relapsed/Refractory Multiple Myeloma with Sequential Addition of the AKT Inhibitor, GSK2141795 at Time of Disease Progression to Overcome Treatment Failure: A Trial of the Princess Margaret Phase II Consortium. Blood 2016; 128 (22): 4526.
-
Schjesvold F, Paiva B, Ribrag V, et al. Cobimetinib Alone and Plus Venetoclax With/Without Atezolizumab in Patients With Relapsed/Refractory Multiple Myeloma. Clin Lymphoma Myeloma Leuk. 2023 Jan;23(1):e59-e70.
-
Costa LJ, et al. Mezigdomide (MEZI) in novel combinations for relapsed or refractory multiple myeloma (RRMM): updated results from the CA057-003 trial. Presented at: International Myeloma Society Annual Meeting; 2025. Abstract.
-
Yang Y, Bolomsky A, Oellerich T, et al. Oncogenic RAS commandeers amino acid sensing machinery to aberrantly activate mTORC1 in multiple myeloma. Nat Commun. 2022 Sep 17;13(1):5469.
-
Norseth HML, et al. The phase I/II TG01 study – vaccinating against RAS-mutated multiple myeloma. Presented at: European Hematology Association (EHA) Congress; 2025.
-
Andrulis M, Lehners N, Capper D, et al. Targeting the BRAF V600E mutation in multiple myeloma. Cancer Discov. 2013 Aug;3(8):862-9.
-
Giesen N, Chatterjee M, Scheid C, et al. A phase 2 clinical trial of combined BRAF/MEK inhibition for BRAFV600E-mutated multiple myeloma. Blood. 2023 Apr 6;141(14):1685-1690.
-
Avet-Loiseau H, Facon T, Grosbois B, et al. Oncogenesis of multiple myeloma: 14q32 and 13q chromosomal abnormalities are not randomly distributed, but correlate with natural history, immunological features, and clinical presentation. Blood. 2002 Mar 15;99(6):2185-91.
-
Kalff A, Spencer A. The t(4;14) translocation and FGFR3 overexpression in multiple myeloma: prognostic implications and current clinical strategies. Blood Cancer J. 2012 Sep 7;2(9):e89.
-
Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood. 1998 Nov 1;92(9):3025-34.
-
Våtsveen TK, Brenne AT, Dai HY, Waage A, Sundan A, Børset M. FGFR3 is expressed and is important for survival in INA-6, a human myeloma cell line without a t(4;14). Eur J Haematol. 2009 Nov;83(5):471-6.
-
Croucher DC, Devasia AJ, Abelman DD, et al. Single-cell profiling of multiple myeloma reveals molecular response to FGFR3 inhibitor despite clinical progression. Cold Spring Harb Mol Case Stud. 2023 May 9;9(2):a006249.
-
Kumar S, et al. Myeloma developing regimens using genomics (MyDRUG) trial: results from the RAS mutation targeting arm. J Clin Oncol. 2022;40(16 Suppl):8055.
-
Landgren O, et al. EVIDENCE meta-analysis: evaluating minimal residual disease as an intermediate clinical end point for multiple myeloma. Blood. 2024;144(4):359–367.
-
Ramberger E, Sapozhnikova V, Ng YLD, et al. The proteogenomic landscape of multiple myeloma reveals insights into disease biology and therapeutic opportunities. Nat Cancer. 2024 Aug;5(8):1267-1284.